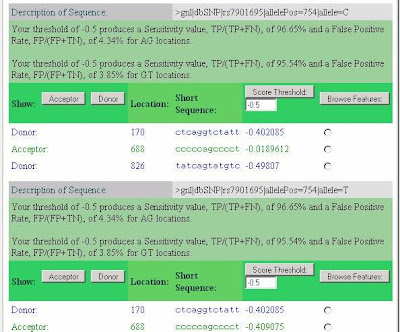
Most striking is the activation of a splice acceptor site 68 nucleotides upstream of the variant SNP (position 688 in the submitted sequence or 114,744,012 on chromosome 10). The SplicePort score, which is -0.41 for the T allele, but -0.02 for the C allele, can be understood by noting that while 95.66% of splice acceptors have a score greater than -0.41, 89.01% of splice acceptors score above -0.02. Thus, the C allele acceptor site, although still relatively weak, is clearly better and well within the range of variation for real splice sites (99% of acceptors score above -0.86 and the median score is 0.923).
How might a C to T change affect an acceptor splice site upstream? Spliceport provides a feature browser that lists the features used for scoring any site. In this case, the following "downstream features" contribute to the score of the C variant but not the T variant: cgg (0.112), ctac (0.083), cg (0.072), ctacg (0.06), tacg (0.059), acg (0.043) and acggg (0.035). An independent approach, ESEfinder, similarly identifies this sequence context (CTACGGG but not CTATGGG) as an exonic splicing enhancer potentially recognized by ASF/SF2, SRp40 or SRp55. Thus, the rs7901695 C variant might activate the upstream acceptor site by functioning as part of an exonic splicing enhancer that is activated by one or more SR proteins.
The next question is how the activation of an acceptor splice site deep within an intron would affect gene expression. A splice site that is activated by mutation is known as a cryptic splice site, and an exon that is used only in mutant alleles is referred to as a cryptic exon. Intron mutations that affect gene expression by creating cryptic exons have been known for some time. In fact, I wrote a commentary on several such mutations in the human beta-globin gene over 25 years ago while still a graduate student ("Lessons from mutant globins," Mount and Steitz 1983). Mutations that activate cryptic exons are often overlooked because they lie away from splice sites, and because the resulting RNA is often unstable due to nonsense mediated decay. Nevertheless, there are now hundreds of papers describing such mutations. The case here is especially tricky because the SNP does not directly create a cyptic splice site, but may activate one at a distance.

Thus, activation of a cryptic exon is a reasonable hypothesis for the effect of the rs7901695 variant on TCF7L2. In this model transcripts from the C allele are more likely than transcripts from the T allele to be aberrantly spliced and ultimately degraded. The lack of EST data supporting a cryptic exon in this region can be explained by nonsense-mediated decay. This proposed mechanism is similar to regulated unproductive splicing and translation ("Evidence for the widespread coupling of alternative splicing and nonsense-mediated mRNA decay in humans." Lewis et al. 2003), an important difference being that cryptic exons are generated by mutation rather than being regulated alternative exons.
How likely is this hypothesis? I could not find any papers that have investigated the effect of this variant on splicing. Clearly, the next step is to look for evidence of the cyptic exon and verify that the C variant does indeed introduce an exonic splicing enhancer. There is also the possibility that other SNPs associated with the risk variant haplotype ("HapBT2D"), particularly rs7903146, are more likely to be causative (Helgason et al. 2007). I could not find a direct comparison of the relative risk for these two variants, but it's possible that association data alone will rule out rs7901695, or even that they already have. Colleagues have suggested that I pursue this in my own lab, but I work other things, and there are people in the diabetes field that can do this quickly. I only ask that they cite this post (here's how).
Although additional investigation is needed, the rs7901695 variant is certainly capable of explaining an effect on the expression of TCF7L2 through activation of a cryptic exon. This case is an example of how SplicePort can be used to evaluate the potential of variants to alter splicing. We plan to systematically evaluate the possible effect of all human SNPs on splicing. In the meantime, I strongly encourage investigators to use SplicePort to evaluate variants of interest on their own.